
Leqembi由卫材和渤健公司联合开发。图片来源:Clear Thoughts Foundation
Lecanemab显著减缓阿尔茨海默病患者认知和记忆功能下降的速度。图片来源:FDA官网
· FDA强调,Leqembi 应该从患有阿尔茨海默病轻度认知障碍或轻度痴呆阶段的患者中开始使用。处方信息指出,在开始 Leqembi 治疗之前应进行 ApoE ε4 基因检测,以了解发生淀粉样蛋白相关成像异常的风险。
·“它真正实现了阿尔茨海默病病理机制上根本性的干预,对全世界阿尔茨海默病患者都有很大的意义。”
Leqembi由卫材和渤健公司联合开发。图片来源:Clear Thoughts Foundation
当地时间7月6日,美国食品药品监督管理局(FDA)宣布,完全批准阿尔茨海默病(AD)新药Leqembi(通用名:Lecanemab)上市。这也是20年来首款获得FDA完全批准的阿尔茨海默病新药。
Leqembi由日本卫材药业(Eisai)和美国渤健公司(Biogen)联合开发,2023年1月,获得FDA加速批准上市。此前的6月9日,FDA外周和中枢神经系统药物咨询委员会(以下简称“FDA咨询委员会”)以6-0投票赞成完全批准Leqembi的决定。
作为加速批准的上市后要求,FDA 要求申请人进行临床试验(通常称为验证性研究),以验证 Leqembi 的预期临床益处。其疗效使用3 期随机对照临床试验Study 301(CLARITY AD)进行评估,Leqembi展示了积极的治疗效果。
“今天的批准首次验证了一种针对阿尔茨海默病潜在疾病过程的药物在这种毁灭性疾病中显示出临床益处。” FDA 药物评价与研究中心神经科学办公室代理主任 特雷萨·布拉奇奥(Teresa Buracchio) 表示:“这项验证性研究证实,它对于阿尔茨海默病患者来说是一种安全有效的治疗方法。”
此次完全获批对Leqembi未来的商业化意义重大,意味着美国医疗保险可以覆盖该药的适应症人群,即阿尔茨海默病轻度认知障碍或轻度痴呆阶段的患者。(详见澎湃科技报道:《阿尔茨海默病药物Leqembi有望获美国FDA完全批准》)
至此,卫材和渤健成功跨越了其早先研发的阿尔茨海默病新药Aduhelm(通用名:Aducanumab)投下的阴影。由于两项设计相同的临床试验未能取得一致结果,Aducanumab在FDA咨询委员会一边倒的反对声中加速批准上市,美国医疗保险和医疗补贴服务中心(CMS)也决定限制其医保覆盖范围,严格限制为仅用于参与临床试验的患者。
和Aducanumab一样,Lecanemab也通过减少大脑中聚集的Aβ发挥疗效。Lecanemab的成功上市可谓对AD发病机制的“Aβ级联瀑布假说”的有力支持,也为靶向Aβ的AD新药开发注入了一剂“强心针”。2022年11月29日,卫材和渤健公布了CLARITY AD 3期关键临床试验数据,证明了Lecanemab的积极疗效。CLARITY AD研究是一项全球多中心、随机双盲、安慰剂对照、平行分组的验证性3期研究,纳入1795例AD源性轻度认知障碍或轻度AD受试者。
Lecanemab显著减缓阿尔茨海默病患者认知和记忆功能下降的速度。图片来源:FDA官网
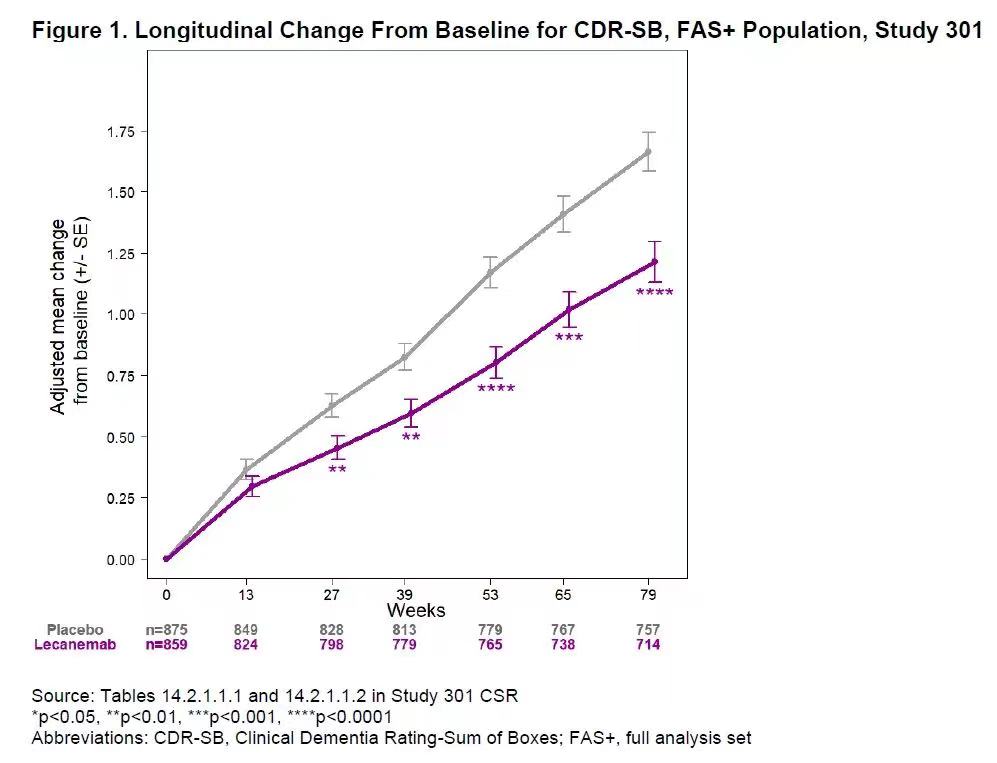
研究结果显示与安慰剂相比,使用Lecanemab治疗18个月后,患者的认知和记忆功能下降的速度减慢了27%,并且在治疗6个月时,用药组和对照组就出现了统计学显著差异。卫材公司曾透露,根据模型推算,与标准治疗相比,Lecanemab可以将早期AD患者进展为中度的时间推迟2-3年,但这一结果还需长期临床试验验证。
在安全性方面,相比其他同类抗Aβ药物,Lecanemab造成的脑水肿和脑出血的发生率相对较低,分别为12.6%和17.3%。该试验也报告了总数不到1%的死亡病例,其中治疗组有6例,安慰剂组有7例,研究者表示,没有死亡病例与Lecanemab相关。(详见澎湃科技报道:《阿尔茨海默病药物Leqembi有望获美国FDA完全批准》)
FDA提到,Leqembi 最常见的副作用是头痛、输液相关反应和淀粉样蛋白相关成像异常 (ARIA)。这是一种已知针对淀粉样蛋白的抗体类会发生的副作用。
“ARIA 最常表现为影像学研究中所见的大脑区域暂时性肿胀,通常会随着时间的推移而消退,并可能伴有大脑内部或表面的小出血点。 尽管 ARIA 通常不伴有任何症状,但可能会出现症状,包括头痛、意识模糊、头晕、视力改变和恶心。 ARIA 也很少会出现严重且危及生命的脑水肿,可能与癫痫和其他严重的神经系统症状有关。 使用此类药物治疗的患者可能会发生脑出血,并且可能致命。 ”FDA称,处方信息中包含黑框警告,提醒患者和护理人员与 ARIA 相关的潜在风险。
此外,接受 Leqembi 治疗的 ApoE ε4 等位基因纯合子患者的 ARIA 发生率,高于该基因杂合子和未携带该基因的患者,包括有症状、严重和重度 ARIA。 处方信息指出,在开始 Leqembi 治疗之前应进行 ApoE ε4 基因检测,以了解发生 ARIA 的风险。(在APOE的3个等位基因中,APOEε4被公认为阿尔茨海默病的最强风险基因。)
与安慰剂相比,服用 Leqembi 的患者使用抗凝药物与脑出血数量增加相关。 处方信息建议,在服用抗凝剂或存在其他脑出血危险因素的患者中考虑使用 Leqembi 时应谨慎。
对 Lecanemab-irmb 或其任何非活性成分严重过敏的患者禁用 Leqembi。 不良反应可能包括血管性水肿(肿胀)和过敏反应。
FDA强调,Leqembi 用于阿尔茨海默病的治疗,应从患有轻度认知障碍或阿尔茨海默病轻度痴呆阶段的患者中开始,这些患者是临床试验中研究治疗的人群。 该药说明书指出,没有关于在疾病早期或晚期开始治疗的安全性或有效性数据。
随着中国日渐成为国际新药研发的重要组成部分,为了补充中国人群数据,由首都医科大学宣武医院牵头,Lecanemab的3期研究也在我国的21家医院开展,第一例受试者于2020年11月10日入组,受试者总数为111人。
澎湃科技记者从卫材公司获悉,这项临床研究已于今年6月6日完成,预计在今年10月初公布结果。2022年12月,Lecanemab已在中国申报上市,最快预计在2024年上半年获批。
近日,澎湃科技记者专访了参与这项研究的首都医科大学宣武医院神经内科主任医师魏翠柏教授,请其解读Lecanemab在临床试验中显示的疗效以及上市的意义。
【对话】
澎湃科技:CLARITY AD中国临床研究进展如何,有没有初步的观察结果?
魏翠柏(首都医科大学宣武医院神经内科主任医师):目前这项研究已经基本“关窗”,今年10月研究报告就会出来。从临床初步的一些观察来看,受试者的反应和症状的改善,包括一些不良事件,应该都跟国外已经公布的数据差不多,中国人群和外国人群的研究结果应该是比较一致的。
澎湃科技:患者招募的过程顺利吗?就你的接触了解,他们在入组前对Lecanemab有怎样的期待?
魏翠柏:Lecanemab是一款对因治疗的药物,在中国的招募比较顺利,比预计时间早了一个多月完成。有很多患者看到医院公众号的通知,就主动找过来想参加这个研究项目。
每个研究中心(参与临床试验的医院)的受试者在10例上下。临床试验方案本身允许患者之前用过一些治疗AD的药物,比如胆碱酯酶抑制剂,但我印象比较深的一个患者,他是首诊,只用了Lecanemab一个药物,在4个多月左右时,我问他感觉怎么样,他说还挺好的,患者自己感觉头脑清醒一些了。
至少这4个月期间患者自觉没有进展,我现在还没有看到患者最后的临床评估结果。这只是个案,不能代表整个群体患者的情况,具体还要等到研究报告出来后,用数据来体现。
澎湃科技:Lecanemab针对的人群是早期患者,他们在入组时的症状如何?
魏翠柏:早期治疗、早期干预符合我们现在对于AD管理的理念。在AD人群中,这些患者属于轻度认知障碍(MCI)或轻度痴呆阶段,症状是偏轻的,典型症状就是记忆力下降,可能伴有一点言语障碍、找词或命名困难等,仅在复杂的日常生活中稍微受到影响,但可以通过提示或他人轻微辅助来完成。
我们现在认为,Aβ是AD的一个致病蛋白,当它累积到一定程度,会出现很多病理性的损害,因此清除Aβ可以一定程度上阻断或逆转疾病的进程。在疾病的早期,有很长的时间窗口可以干预,Lecanemab的研发就是针对这个倾向,越早清除Aβ,后期治疗效果可能更好。就Lecanemab的作用来说,它对于清除毒性更强的Aβ寡聚体和原纤维,都有一定效果。
澎湃科技:过去靶向Aβ的药物临床试验失败,是否就和患者大脑中已经沉积过多的Aβ有关?
魏翠柏:这是一个很复杂的问题,药品研发失败不一定取决于患者疾病进程较晚。我们目前一线用的药物胆碱酯酶抑制剂、美金刚用于治疗轻度、中度、重度患者一样有效,但它不是针对疾病病因本身的,而只是改善症状。所以一个药品研发是否成功,主要看它的适应症人群和作用机制,对因治疗和对症治疗的目的和期待是不同的。针对不同药物的特点,可以选择不同人群,用不同观察指标去评价。
澎湃科技:Lecanemab在中国患者中使用的安全性怎么样?
魏翠柏:从不良反应事件的发生率看,中国和国际已公布的试验数据差不多。大家最关心的淀粉样相关影像学异常,包括脑水肿和脑出血,在我们医院的11名受试者里,我个人感觉发生率并不高,暂时不知道全国整体数据。临床试验中对不良反应事件的处理,有一套标准化模式,是国际通用的。
澎湃科技:中国研究中心对CLARITY AD研究的意义是什么?
魏翠柏:全球多中心临床试验采用统一的临床方案,目的就是看这种药物对于不同种族、地域的人群,是否具有相同的效果。我们的研究就贡献了中国地区数据,观察了在中国人群特定遗传基因背景和生活模式下,药物的效果和安全性,对药物在全球的效果做了补充和验证。有了中国患者的数据,也会更好地支持这个药在中国的上市。
澎湃科技:你怎么看Lecanemab上市对患者的意义?
魏翠柏:Lecanemab对全世界AD患者的意义都很大,因为它真正实现了我们一直关心的从疾病病理机制上根本性的干预。1998年后,第二代胆碱酯酶抑制剂安理申上市以来,还没有特别突破的药物,我觉得像Aducanumab和Lecanemab从一定意义上来讲打破了这种僵局,真正从病因的机制本身去开发了药品,抑制这种疾病的根本性致病作用的一些关键的靶点,来做干预。所以,Lecanemab上市是阿尔茨海默病治疗中一个里程碑性的突破,当然我们期望它更有效、更安全。

·美国一直是新药支付最慷慨的市场,可如果连欧洲市场都不愿意支付,那Lecanemab的全球商业前景无疑会黯淡一些。美国市场足以支撑起Lecanemab的重磅新药梦,但如果“钱”途完全限于美国,加上前面提到的适用人群、安全性等不确定因素,商业风险无疑在增加。
2023年7月6日,FDA(美国食品药品监督管理局)将渤健/卫材的阿尔兹海默病新药Lecanemab的加速批准转为正式批准。这一转变有重大意义,因为这意味着Lecanemab的商业化将正式起步,备受关注的β样淀粉蛋白抗体药将开始真正的“上市”。
Lecanemab在2023年1月获得FDA的加速审批上市(accelerated approval),成为继Aducanumab后第二个获FDA批准上市的阿尔兹海默病治疗药物。
过去的阿尔兹海默病药物都是针对症状缓解,而Aducanumab和Lecanemab这类β样淀粉蛋白抗体,作用机理是清除阿尔兹海默病患者大脑里的β样淀粉蛋白斑块,希望以此来缓解疾病恶化,是真正治疗性质的阿尔兹海默病药。
但在FDA今天完全批准Lecanemab前,两个上市的阿尔兹海默病药从商业角度只能算是“伪”上市。
这是由于阿尔兹海默病患者主要是老人,在美国,他(她)们主要使用Medicare保险(联邦医疗保险)。Aducanumab获得加速审批的过程争议极大,又会给Medicare带来极大的经济负担。于是Medicare在Aducanumab上市后干脆搞了个特殊政策:不是全面获批的β样淀粉蛋白抗体药,只付临床试验里的药物费用。
该政策让Aducanumab经历了医药史上最迅速的从上市成功到上市失败的转变。Aducanumab凭借清除了β样淀粉蛋白板块这一生物标记物获得的加速审批,没有展现出明确的临床有效性数据(两个三期临床试验,失败了一个,另一个仅高剂量组显示有效),没有全面获批,等于被Medicare拒了。
FDA当时以生物标记物指标变化批准Aducanumab加速上市争议很大,最关键的一点:谁也不敢说这药真有效。Medicare限制支付的理由就是:咱还是先搞明白有没有效,再付钱不迟。
但Aducanumab的所有者——渤健/卫材其实没太大兴趣去搞清楚Aducanumab到底有没有效。这倒不是有什么阴谋论,而是它们的下一个β样淀粉蛋白抗体,Lecanemab三期临床试验已经开始,重新去做Aducanumab的试验,不仅投入巨大,出数据时间还肯定会远晚于Lecanemab,甚至要晚于礼来等竞争对手的β样淀粉蛋白抗体。
Medicare的限制实际上宣判了Aducanumab的死刑,加上该药物晚期临床试验结果的不清不楚,也成了β样淀粉蛋白抗体药物头上的一朵乌云。但Lecanemab后来的数据让整个β样淀粉蛋白抗体领域一扫阴云:三期临床试验中延缓认知衰退27%。
也就是说,Lecanemab不仅清除了β样淀粉蛋白斑块,这一阿尔兹海默病患者的经典病理学特征,还确实延缓了疾病进展,有临床有效性。
而临床有效性就让Lecanemab有了被FDA全面批准的资格,不会只能依靠加速批准。Medicare的支付限制也很明确:获得全面批准的β样淀粉蛋白药物不会受限制。
因此,Lecanemab也就成了能“真”上市的阿尔兹海默病新药。但Lecanemab也在临床试验中证实了它能清除β样淀粉蛋白斑块,符合Aducanumab时FDA立下的加速审批规则,于是2023年1月,Lecanemab成了第二个加速审批上市的β样淀粉蛋白抗体。
注意,和Aducanumab一样,Lecanemab此时是加速审批,证据是可以清除β样淀粉蛋白斑块,因此,按Medicare之前定的规矩,得等到获得FDA正式批准才能获得广泛的支付支持。可和Aducanumab不同的是,Lecanemab当时已经有了明确阳性的三期临床试验结果,只是这些有效性安全性数据还未经过FDA正式审核认可。不支付的决定让很多人觉得Medicare这是不让患者用上有效药。等于Medicare又被逼到了非常尴尬的位置。
挺逗的是,Aducanumab获批时,由于预计Medicare支付该药物需大量资金,美国当时大幅增加了老人需要支付的Medicare保费,彼时通胀问题正成为全美最受关注的议题,保费上涨无疑会让民众怨声载道,也让政府焦头烂额。加上Aducanumab自己数据不够争气以及获批时FDA种种过度积极的绿灯,让这种Aducanumab通胀成了美国政界的出气筒,多位议员公开表示Medicare不能为它买单。美国卫生部长更是建议负责制订Medicare保费的机构先别涨价,表示Medicare买不买单还不知道,等确定买单了咱再涨价也不迟。
等到Lecanemab这会儿,同样是加速审批,但有了有效性数据撑腰,又变成另一波议员向Medicare施加压力,要求支持支付。不过Medicare最后还是决定秉承自己立下的规矩,以生物标记物为标准,加速审批上市的阿尔兹海默药仍然限定支付范围在临床试验里,只有获得FDA正式批准后才撤除上述支付限制。
由于有明确的支付限制,Lecanemab获得加速审批上市后,渤健/卫材极为低调,一改Aducanumab获批时要大干一场的豪气,甚至直言在正式获批前不会做商业扩展努力。
不过,Aducanumab获批时的豪情多少带着心虚,想以造势的方式压过多方质疑。此时“安静”的背后其实是对Lecanemab的数据极有信心:既然正式获批只是时间问题,而Medicare支付到时会迎刃而解,就不必冒险去趟混水。毕竟阿尔兹海默病药物巨大市场的另一面就是极有争议的巨大支付负担,药企冒然施压,不仅未必有啥用,还很容易被指责贪婪、试图影响监管独立性。而且FDA预计的决定时间就是2023年7月,加速审批上市后半年不到,更不值得去节外生枝。
2023年6月9日,类似Aducanumab上市审核时的做法,FDA为Lecanemab召集了外部专家听证会。由于Lecanemab数据远比Aducanumab清晰,该听证会也就和当年Aducanumab听证会上剑拔弩张的态势成了鲜明对比。最终,外部专家全票同意Lecanemab显示了临床有效性,为其上市扫清了最后的障碍。
如今Lecanemab获得正式批准,成了第一个获得FDA正式批准的阿尔兹海默病药物。这意味着Medicare很快将开始为Lecanemab买单。由于大量的阿尔兹海默病患者人群,Lecanemab等药物一直被预计会是重磅药物,很快我们也能看看这是否能成为现实,以及需要多久才能实现。
由于受阿尔兹海默病困扰的患者极多,很多人一方面对Lecanemab这类新药充满好奇,另一方面也会想这类药物到底作用会怎么样。
尽管对于多年来没有突破的阿尔兹海默病新药研发来说,Lecanemab展示出来的有效性是重大进展,但在实际应用上,对这类药物还是要有理性的预期。
首先,只有少数阿尔兹海默病患者会适用lecanemab这类药物。只有在存在β淀粉样蛋白斑块的早期(症状轻微)患者中,Lecanemab才能通过清除β淀粉样蛋白斑块来延缓疾病进展。两个前提缺一不可,有β样淀粉蛋白斑块,Lecanemab才能做到有的放矢;同时,只有在症状轻微阶段使用才能有延缓病情恶化作用,失智症已经严重了,用此类药物就没太大意义。
其次,Lecanemab有不可忽视的副作用。β样淀粉蛋白抗体药是激活脑部的免疫细胞来清除斑块,这种机理意味着容易在脑部引发炎症,有导致一种叫ARIA的副作用。ARIA表现为脑部成像的异常,背后可能是出血或水肿,严重时可能会导致死亡。
再次,需要明确β样淀粉蛋白斑块的存在以及ARIA都意味着要有效、安全地使用Lecanemab,需要极为复杂的支持系统。其中最关键的就是能经常获得脑部医学成像检查的机会——既确定斑块的存在,保证是适用人群,又来排查监测ARIA,及时处理控制这类危险的副作用。这对于很多患者以及很多地区来说并不现实。
上述因素也让Lecanemab如今“真”上市后,实际商业扩展能否顺利存在一定的不确定性。
这些属于科学因素,还有其它因素也会影响Lecanemab的未来。一个是全球支付方对这类药物的认可程度,目前看来美国由于Medicare已经首肯,问题不大。但6月,欧洲的阿尔兹海默病专家组认为Lecanemab的有效性有限,不具备充分的价值。
美国一直是新药支付最慷慨的市场,可如果连欧洲市场都不愿意支付,那Lecanemab的全球商业前景无疑会黯淡一些。美国市场足以支撑起Lecanemab的重磅新药梦,但如果“钱”途完全限于美国,加上前面提到的适用人群、安全性等不确定因素,商业风险无疑在增加。
此外,Lecanemab在不远的将来还会迎来一个有力的竞争者:另一个β样淀粉蛋白抗体药——礼来的Donanemab。5月初,礼来宣布Donanemab在三期临床试验中也显著延缓了阿尔兹海默病患者的疾病恶化。由于试验设计不同,两个药物目前很难区分有效性差别,整体来说可能接近,都属于在轻微症状患者中使用两年,大概延缓半年疾病进展。
Donanemab的优势是注射间隔更长,每四周注射一次,Lecanemab是两周一次注射,患者负担更大,是显著劣势。但另一方面,ARIA在临床试验中的比例,Donanemab要高一些,安全性问题上可能会处于劣势。
两大类ARIA,ARIA-E和ARIA-H。根据礼来公布的数据中,ARIA-E在Donanemab组的发生率是24%,有症状的ARIA-E是6.1%。而在Lecanemab的三期临床试验中,ARIA-E发生率是12.6%,有症状2.8%。ARIA-H,Donanemab发生率是31.4%,而Lecanemab三期临床试验里是17.3%。
如今Lecanemab正式上市,也获得了针对Donanemab最宝贵的优势:时间。
Donanemab预计2023年上半年完成上市申请,几乎不太可能在今年获得正式批准。Lecanemab能否把握住宝贵的“独占”期,让医生、患者以及支付方都能认同其价值,很可能会决定它能否压制住Donanemab。
但另一方面,Donanemab的成功很可能也会“依赖”于Lecanemab。如果医生、患者以及支付方中任何一方实际对Lecanemab不感冒,导致其商业扩展困难,很难想象Donanemab上市后就会被认可。
对于阿尔兹海默病药物的未来来看,短期内可能也就只有Lecanemab和Donanemab两个药物。整体来看,β样淀粉蛋白抗体药物可以一定程度上延缓疾病进展,也就是具备有效性,但有效性不算高,适用人群也很有限。进一步突破可能需要依赖不同机理的药物,而非吊死在β样淀粉蛋白这一棵树上。
重复做一个β样淀粉蛋白抗体去和这两位先行者竞争更是意义有限。毕竟这两个已经是很好的β样淀粉蛋白抗体了,极为有效的清除β样淀粉蛋白斑块就是力证。改善的空间或许在于抗体这类大分子药物穿过血脑屏障进入大脑的效率极低,导致Lecanemab和Donanemab的使用剂量、频率都极高,这既带来了使用不便、更容易出现注射相关不良反应,又让生产成本高昂。如果在大分子药物递送上有突破,可以更有效穿过血脑屏障,那么或许能出现一个剂量更温和,生产成本也大幅降低的β样淀粉蛋白抗体。
Lecanemab剂量高达每千克体重10毫克,每两周注射一次,对抗体药来说属于“海量”。如今美国定价约2.6万美元一年,考虑到用药量,不能算黑心,但阿尔兹海默病如此大的患者基数,这类药物也只能适合少数支付更慷慨的市场的奢侈品。
以中国市场为例,这样的高药价对于一个患者基数极大的疾病来说,肯定难以接受。另一方面,这类药物使用的复杂性——频繁的注射、脑部成像监测的要求,也意味着它们缺乏必要基础设施支持。
但作为近年来最为重大的一类新药,其开拓全新的治疗领域的过程,如何向医疗工作者、患者介绍普及这类药物的作用,如何应对较高的医疗基础条件要求挑战,都非常值得关注。
这也是为什么Lecanemab的“真”上市值得关注。
(作者周叶斌,系美国阿拉巴马大学伯明翰分校遗传性博士,长期从事免疫学研究,目前在药企从事新药研发。本文首发于“一个生物狗的科普小园”微信公众号,澎湃科技获作者授权刊发。原标题《开启“真”上市之路:阿尔兹海默新药lecanemab获得FDA全面批准》)